Biology, Genomics, and the Information Age
Author’s Note: In my last year of high school, I had the privilege of conducting bioinformatics research, working with amazing people, and eventually publishing it in Nature Communications. I get asked all the time about this research–and have found it quite difficult in the past to paint a unified tapestry illustrating recent scientific history while accessibly describing current work on the 3D genome. I wrote this piece to describe this field of work to a layperson, with a particular focus on the central role that informatics has taken in biology. This piece was originally published as a featured article in the Harvard Science Review.
Introduction
Imagine a charging cable, six feet long, stretching from arm to arm.
Now imagine the same cable, fully extended, width shrunk 2 million times—from 5 millimeters in diameter to 2.5 nanometers. In your hands now is the world’s most precise nano-manufactured cable, a tensile string held taut by powerful atomic forces and quantum flux: the DNA from a single human cell.
What makes this image so outlandish is that it is true. If fully extended, the DNA inside of a human cell would stretch six feet. Inside of a cellular nucleus, however, it is packed into a space that is just one-tenth the width of a human hair. Within that long string lies a code, an intrinsic human programming language of 3 billion tightly-strung chemical nucleotides: A’s, T’s, G’s, and C’s.
Compressing a string that is six feet long into such a tiny space is no easy task, it turns out. Inside of the nucleus, the strand tightly coils and weaves into a spaghetti ball of DNA, wrapped by tiny sheaths of protein and fiber that keep the entire edifice intact. Inside the nucleus, billions of complex chemical interactions guide the actual regulation and expression of our DNA, which is separated into several hierarchies of organization.
In this feature, we’ll first dive into the history of modern genomics to understand the frontiers of the field, where the edges of our understanding meet vast swaths of what is still unknown. We’ll see why getting your DNA sequenced in a kit isn’t enough to understand your genome, and what other new assays are being developed to probe what is now known as the “3D Genome.” Finally, we’ll see how modern cellular biology is driven by informatics—-and how the triumph of computational power has powered much of modern genomics.
From One-Dimensional Sequence . . .
In many ways, 1953 was the start of the modern genomics revolution.
In the decades prior, researchers had made a series of curious—but seemingly disconnected—discoveries. The chemist Alexander Todd, for instance, determined that DNA contained repeating phosphate and deoxyribose sugar groups. Erwin Chargaff, a biochemist, worked out that DNA is composed of four purine and pyrimidine bases: adenine (A) and guanine (G), cytosine (C) and thymine (T).
In 1952, however, the now-famous duo of James Watson and Francis Crick (using work by Rosalind Franklin and Maurice Wilkins) announced the unifying structure behind DNA: the double helix.1
While their article, a one-page note in Nature, garnered scant attention on publication, research over the following decades soon catapulted it back into the spotlight. Driven by the dream of deciphering what Watson described as “the code of life,” researchers on both coasts of the U.S. began developing methods to parse the letters in small fragments of DNA. At Harvard, Walter Gilbert developed the first assays for rapid DNA sequencing; in Silicon Valley, the budding informatics revolution finally offered the computational scale necessary to parse millions (and soon billions) of data points. By the late 1980s, many wondered if we finally had the resources for “the holy grail of human genetics.” Could we decipher the chemical totality of what makes up humanity—and parse the complete human genome?
The answer, as we all know, is yes. In 2001, following a decade-long, $2.5 billion effort, the Human Genome Project (HGP) announced the first-ever complete draft sequence of the genome.2 3
Expectations for the HGP were high. Since our DNA encodes every biochemical process in our body, many believed that it was only a matter of time before we could “solve” every human complexity by working purely off the linear sequence. In many ways, the vision was seductive: on-demand genome sequencing, backers claimed, would lead to a whole new era of personalized medicine, one where anyone could take one test and instantly see their precise risk factors for disease, susceptibility to different drugs, and even fertility metrics. Indeed, when President Clinton and future director of the National Institutes of Health (NIH) Francis Collins announced the first draft of the human genome in 2001, they described it as the first step toward a day when “our children’s children will know the term cancer only as a constellation of stars.” 4
. . . To Three-Dimensional Architecture
Today, that original vision for the HGP seems largely like a pipe dream. Although complete genome sequencing is here and can be done at costs under $1000, many of the promises of the HGP seem further away than ever. This is due in large part to many of the discoveries made during and through the HGP, advances that have helped us unlock the next piece of our biological puzzle: the three-dimensional structure of DNA.
While the linear sequence of base pairs that make up DNA encodes things like genes, promoters, and enhancers, most genomic function occurs because of factors “beyond the line”-—elements that guide the function of DNA that are not in its raw encoding. Today, we call the field that explores those genomic processes epigenomics, the study of elements that are epi, in addition, to the linear sequence of A’s, G’s, T’s, and C’s.
To see how this takes place, we consider a prototypical example of DNA looping: the Sonic Hedgehog (Shh) gene. A gene whose expression is critical for developmental morphology across many species, Shh’s expression is regulated by a variety of enhancers that span a 900 kilobase pair (900,000 nucleotides) linear swath of the genome. In other words, the necessary machinery for this gene lies on a long linear stretch of DNA.
When Shh is expressed, however, the enhancers and promoter must colocalize to form a proximal interaction in 3D space.5 Since the necessary machinery for this gene lies far apart in a linear sequence, the physical DNA containing the machinery must fold to bring the components together. Indeed, as we’ll later see, the Shh domain is a “topologically-associated domain” (TAD for short): a special unit of genomic organization discovered in the past decade.
In a real sense, the topology of DNA shapes its expression: distant regulatory elements are brought together in the three-dimensional folding of the genome.
Mapping Interactions in 3D Space
While the example of Shh interactions above was discovered through thousands of man hours spent carefully sequencing and mapping the local structure of DNA via x-ray crystallography, this process is intractable at a larger scale.
One common assay to map the shape of DNA in 3D space is Hi-C, an enormously popular assay used to generate maps of genomic interactions.6 To see how it works, let’s return to the spaghetti analogy, which offers a crude parallel to what happens inside of a cellular nucleus.7
Like spaghetti in a pot, DNA is in constant flux (Figure 1A). The first step of the Hi-C protocol is “flash-freezing” that spaghetti at some moment in time, “gluing” together spaghetti noodles where they are touching (this is done using formaldehyde, which crosslinks DNA in 3D proximity). Then, using a restriction enzyme, it “snips” away those glued connections, each of which is composed of either two distinct parts of one noodle (an intra-chromosomal interaction) or parts of two distinct noodles (an inter-chromosomal interaction). Finally, those snipped pieces, or “paired ends,” are then sequenced (which tells us which genomic locus each snip is from). See Figure 1B for the entire process.
To visualize the 3D interactions between loci in one chromosome, one can envision an x-y matrix where each axis represents the loci in one chromosome, and the value at point (x, y) represents some level of “interaction” between those loci. Having a matrix of interactions drastically increases our “search space” of interactions and offers a succinct visualization of 3D interactions (see Figure 1C below). These “Hi-C matrices” are ubiquitous in the 3D genomics research field.
Figure 1
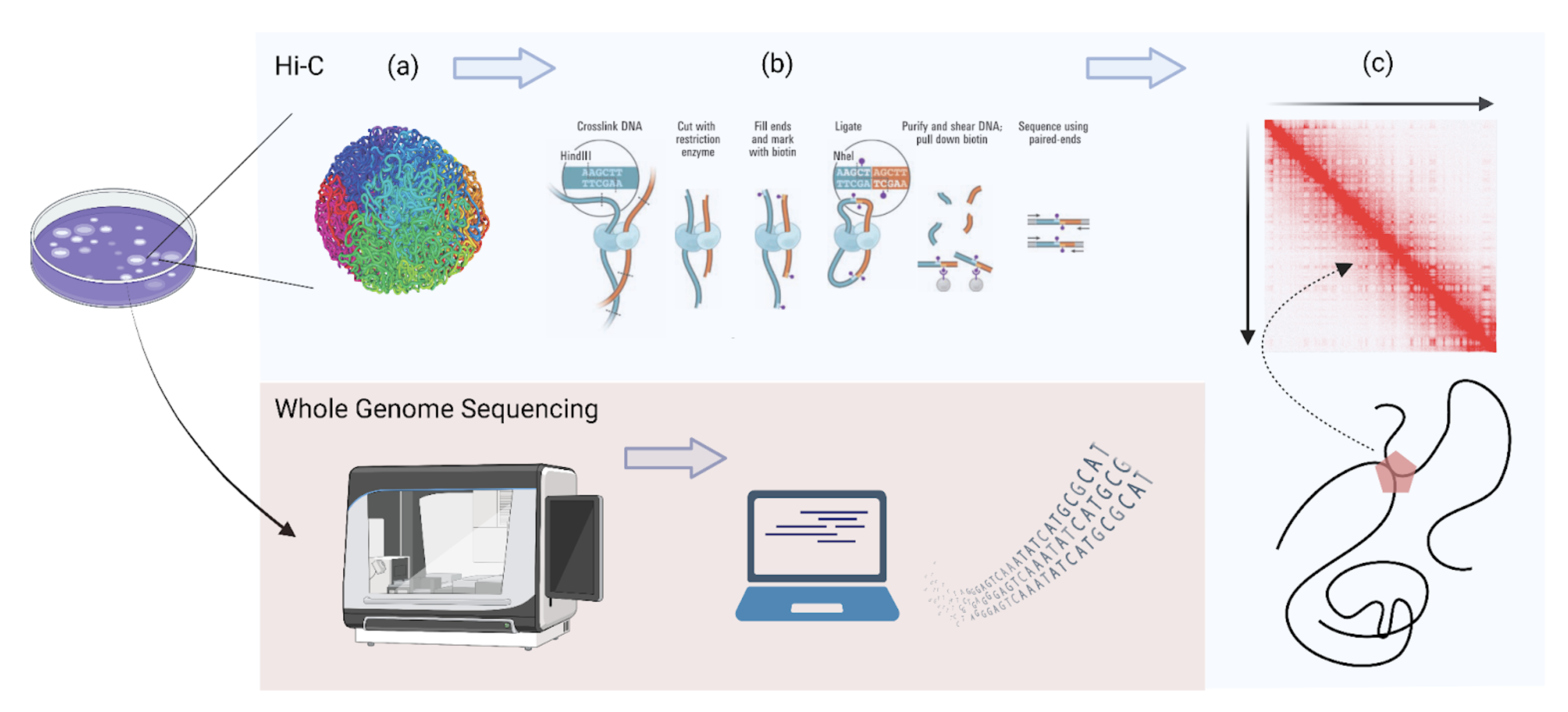
Caption: Hi-C vs Whole Genome Sequencing (WGS). While WGS simply maps the linear sequence of the genome (red box), Hi-C captures an intuitive three-dimensional view (blue). As one would expect, Hi-C matrices are symmetric; that is, they are mirrored along the diagonal (since the 3D interactions between points (x, y) in the genome is the same as the 3D interactions between points (y, x)). Figure created by author.
Figure 2
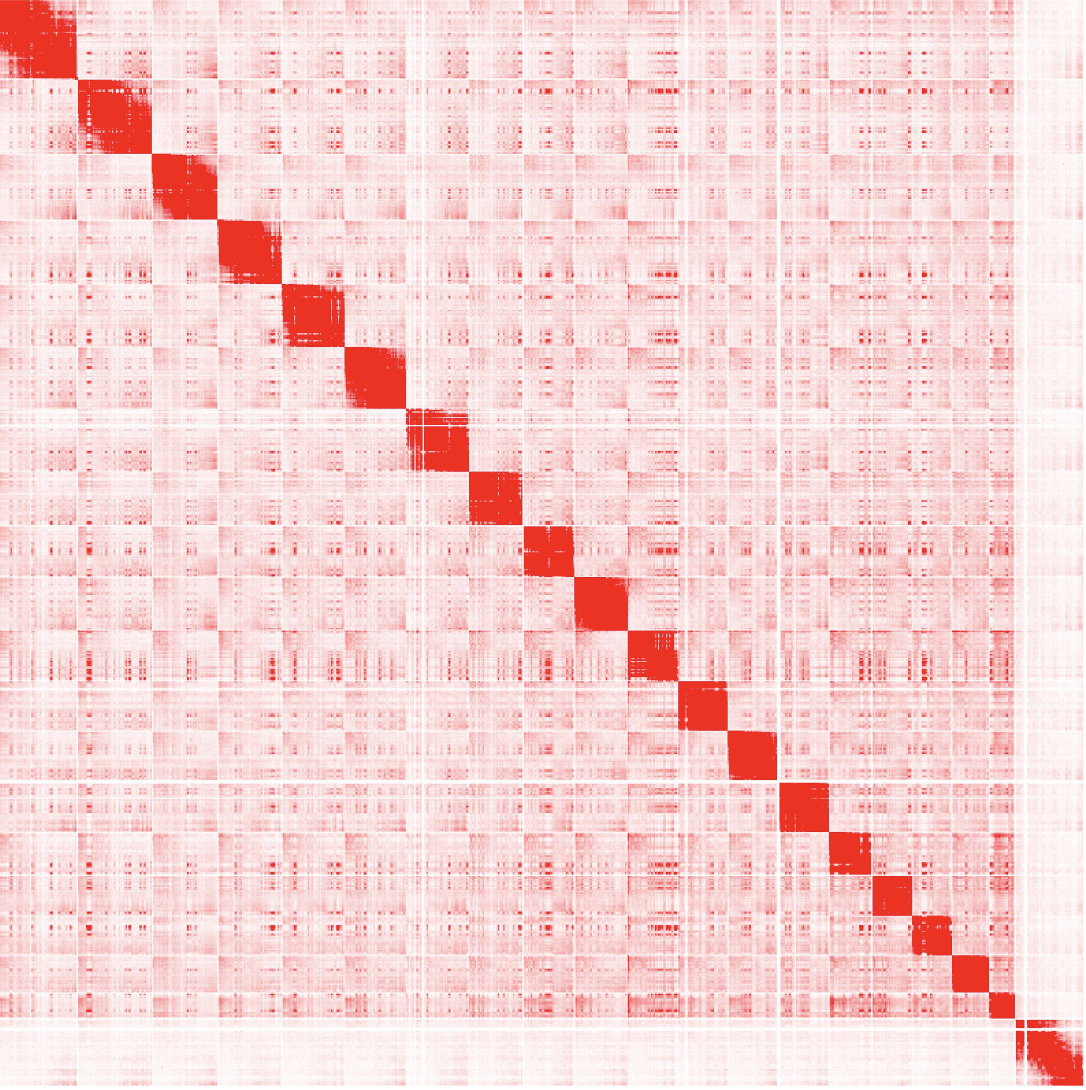
Caption: A genome-wide symmetric Hi-C matrix of a mice embryonic STEM cell. Each row/column delineation represents a chromosome. Figure used with permission from the lab of Ferhat Ay (author’s lab).
You’ll notice in Figure 1C that a standard intra-chromosomal Hi-C matrix is heavily enriched for interactions on the diagonal. This makes sense from a strict distance perspective; areas of DNA that are close in 1D space are more likely to be close in 3D space as well. Tessellated along the middle with each chromosome’s Hi-C matrix, the genome-wide matrix is similar; 3D interactions within one chromosome are simply more prevalent than between chromosomes (Figure 2).
As we’ll soon see, however, it’s the grid-like pattern of these matrices—particularly the off-diagonal interactions—where the true biological magic happens.
Consider, for instance, two common (but catastrophic) genomic events: chromosomal translocations and chromothripsis. The former occurs when a chunk of one chromosome exchanges with a chunk of another chromosome; one common example is the Philadelphia Chromosome, a translocation between chromosome 9 and 22 that is present in about 30% of adult lymphoblastic leukemia cases.8 The 3 left Hi-C matrices in Figure 3 below illustrate examples of translocations, where the balanced matrices of chromosome 1 and chromosome 2 exchange genetic material. The chromosomal breakpoints are circled in red.
Chromothripsis is a mega-scale analog of translocations: one can quite literally think of it as a chromosomal explosion. A single catastrophic event, chromothripsis occurs when a chromosome shatters into many genomic fragments, which spontaneously and stochastically recombine into a nonsense genetic encoding. While the mechanisms of the chromothripsis are still poorly understood, it has been implicated in 49% of adult human cancers.9 10 11
The far right matrix in Figure 3 illustrates chromothripsis in a Hi-C matrix: examine it carefully to see the scattershot off-diagonal enriched interactions: those are genomic fragments that have broken off elsewhere and recombined in odd areas.
As we saw with Shh, the 3D structure of the genome directly influences downstream expression. It comes as no surprise, then, that major rearrangements in the packing of the genome can cause major changes in cellular function (i,e. cancer).
Figure 3
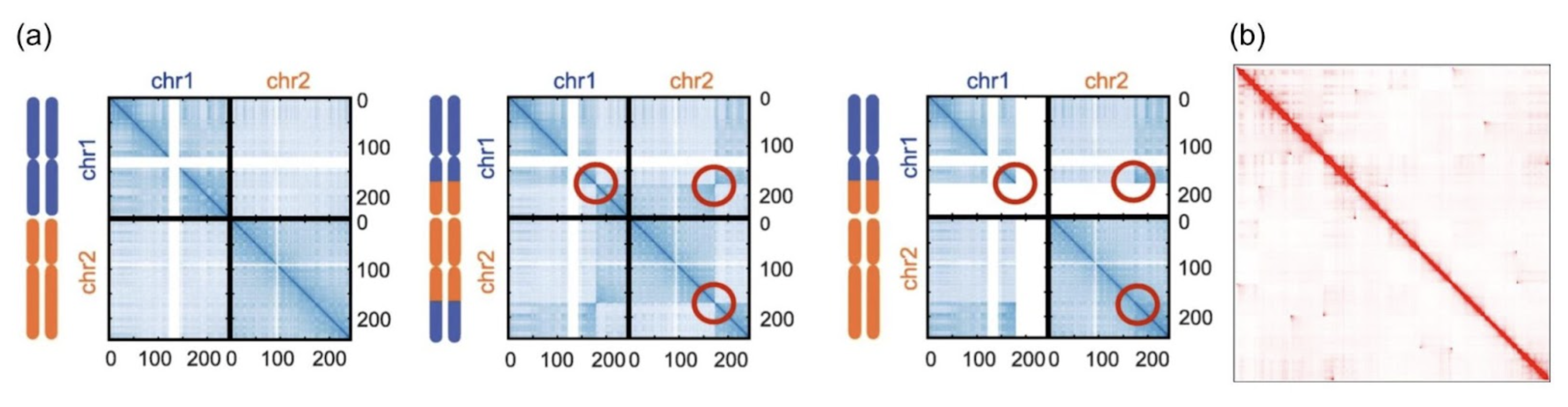
Caption: (A) Translocations, and (B) Chromothripsis. (A) used with attribution from [12] and (B) used with permission from the lab of Ferhat Ay (author’s lab).
Translocations and chromothripsis are extreme examples, but they illustrate the intuitive distillation that a Hi-C matrix offers. Hi-C, in a very real sense, allows us to look “beyond the line” of one-dimensional DNA.
While Hi-C only provides a snapshot of a cell’s nuclear structure at one given moment of time, weaving together the shots allows us to form a rough vignette of the complex process being studied. For instance, to understand the molecular dynamics in lung cancerogenesis, we may wish to compare the Hi-C map of a healthy lung epithelial cell with one that is cancerous. To understand what occurs when a stem cell differentiates into a blood cell, we may take a time series of Hi-C snapshots throughout the process.
Probing the genome isn’t just limited to mapping DNA. Outside of proximal DNA interactions, many other biological elements guide DNA function, each measurable by a different series of assays; ChIP-Seq for DNA-protein interactions, ATAC-Seq for chromatin accessibility, RNA-Seq for gene expression, and others.12
Unsurprisingly, there is a wealth of insight hiding in these datasets: the latest high-resolution datasets generated are hundreds of gigabytes in size. Hidden amidst the enormous binary encodings lay real biological insight; it is simply up to the skilled bioinformatician to extract the signal amidst the noise. In many ways, this is the central challenge of modern bioinformatics: we have collected entire hard drives worth of data, yet still only have scratched the surface of the mechanisms that drive epigenomics.
The Triumph of Informatics
As a result of quantum leaps in the capacity and speed of data collection in the past two decades, central questions in cellular biology have transformed from scientific to informatic in nature.
Although it took 13 years, sequencing the first human genome spurred enormous technological breakthroughs, from new alignment and assembly algorithms to automated DNA sequencers and sample preparation instruments. Since then, we’ve effectively cubed our search space by allowing ourselves to analyze the genome in three-dimensional space.
The shift toward data-driven genomics solutions reflects a central interplay between biology and informatics in modern research. As we generate data, we formulate new questions and develop methods to answer them; this process leads to more data, which leads to more questions, and the cycle continues.
Because of data’s central role in modern cellular biology, the need for standardized computational methods has similarly expanded. This body of research builds computational tooling around the biology of the assays themselves, with an aim of identifying biologically relevant features given any dataset, across tissue or organism. Within Hi-C alone, there are dozens of labs developing computational packages for a gamut of interesting problems: identifying significant DNA interactions, identifying structural genomic features, creating intuitive visualizations, and more.13 14 15 16 17
Before the last several decades, computation usually played second-string to wet-lab biology; questions were resolved experimentally when possible and computational methods were created to address specific problems in that process. Today, however, many labs are computational-first. Many don’t even produce their own data; instead, they identify open problems in the field and leverage deeper technical expertise to create solutions.
To be clear, questions certainly remain. Some are philosophical: within the basic sciences, there has been a long distrust of relying on pure informatics to inform discovery. Mathematically-minded readers may remember the Four-Coloring Theorem, the first major theorem proved using a computer-assisted proof. While the proof is broadly accepted today, it evoked furor when first announced; the use of computational verification seemed to betray the central principles of a field based on constructing results from first principles. Other questions are instrumental: when we struggle to resolve key questions, is it the biology or the computation that is letting us down? So many assumptions go into the discrete steps of collecting data and then analyzing it that it can be difficult to discern which causes the underlying problem when things go wrong.
Despite these concerns, biology has seen an unprecedented expansion in the last few decades. The march toward a computation-first world has accelerated our pace of discovery and increased accessibility, opening the field to huge new swaths of talent. With each additional compute node used for analysis and new algorithmic optimization made, the hypothesis search space grows ever wider, and the data net we cast becomes ever larger.
In biology, it’s the information age. With our quiver of computational tools, we will be able to dive ever deeper into the mysteries of the genome.
Acknowledgements
I’d like to acknowledge my lab mentors, Abhijit Chakraborty and Ferhat Ay, for guiding my research in the 3D genome space. I’d also like to thank everyone at the La Jolla Institute for Immunology for providing a wonderful home to this research.
References
-
Watson, J., Crick, Nature. 1953, 171, 737–738. ↩︎
-
Lander, E. S. et al. Nature. 2001, 409, 860–921. ↩︎
-
Venter, J. C. et al. Science. 2001, 291, 1304–1351. ↩︎
-
The White House, Human Genome Project Press Conference. https://www.genome.gov/10001356/june-2000-white-house-event. ↩︎
-
Jeong Y, et al. Development. 2006, 133, 761-72. ↩︎
-
Lieberman-Aiden E, et al. Science. 2009, 326, 289-93. ↩︎
-
Rao, S. S. et al. Cell. 2014, 159, 1665–1680. ↩︎
-
Kang ZJ, et al. Chin J Cancer. 2016, 35-48. ↩︎
-
Maher CA, Wilson RK. Cell. 2012, 148, 29-32. ↩︎
-
Voronina N, et al. Nature Communications. 2020, 11, 2320. https://www.nature.com/articles/s41467-020-16134-7 (accessed Nov. 2022). ↩︎
-
Cortés-Ciriano, et al. Nature Genetics. 2020, 52, 331–341. ↩︎
-
Kempfer, R., Pombo, A. Nature Reviews Genetics. 2020, 21, 207–226. ↩︎
-
Kaul A, et al. Nature Protocols. 2020, 15, 911-1012. ↩︎
-
Roayaei Ardakany, et al. Genome Biology. 2020, 21, 256. ↩︎
-
Chakraborty, A., Wang, J.G. & Ay, F. Nature Communications 2022, 13, 6827. https://doi.org/10.1038/s41467-022-34626-6 (accessed Nov. 2022). ↩︎
-
Bhattacharyya, S., et al. Nature Communications 209, 10, 4221. https://doi.org/10.1038/s41467-019-11950-y (accessed Nov. 2022). ↩︎
-
Chakraborty A, Ay F. Bioinformatics. 2018, 15, 338-345. ↩︎